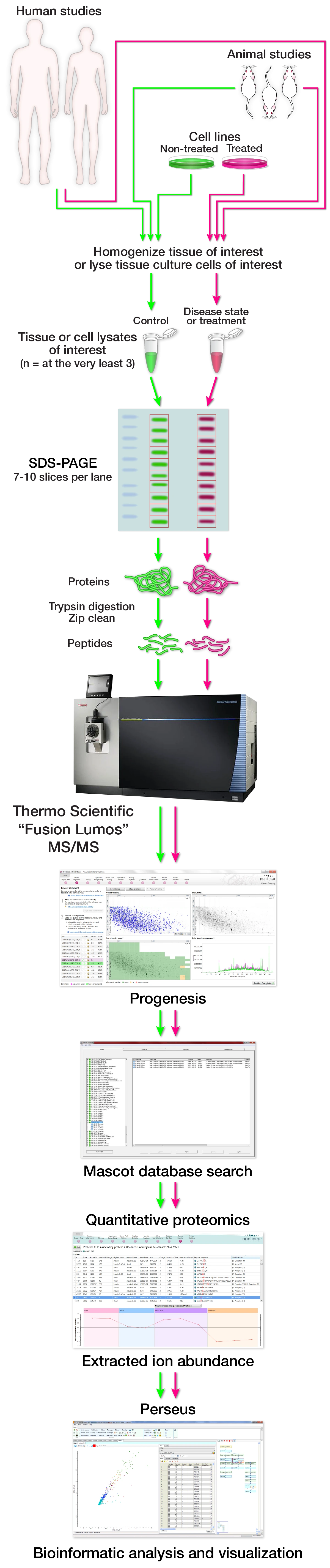
Profiling protein abundance changes across entire proteomes, from either cell culture systems, animal models, or human studies. Since we have some amazing instruments, we are able to get pretty solid depth of proteome coverage that is competitive in the current field of proteomics.
If you have a hypothesis that a certain biological perturbation results in global protein expression changes across entire proteomes, we would like to help. It is our responsibility to aid in the design and execution of the experiment. It begins with a conversation of the specifics and discussion of feasibility, potential complications, and hopeful outcomes. After that, due diligence is performed to ensure efficient proteome coverage. Once appropriate proteome coverage has been validated a decision is made on the final experimental design and cost. Protein lysates (anywhere from 25-200ug) from cell culture or animal/human tissue are separated by SDS-PAGE and each gel lane is then fractioned into the chosen number of gel slices. Each gel slice is then prepped, subjected to enzymatic digestion to digest the protein into peptides, and the resultant peptides are then desalted. Each gel slice's purified peptides are then analyzed as independent samples by mass spectrometry. The mass spec raw data is then processed in the quantitative proteomics software program Progenesis followed by searching against the appropriate database with the software program Mascot. The Mascot peptide and protein identification data is then imported into Progenesis. The proteins are then analyzed for expression changes across the different treatment groups. The proteomic data can then be imported into the bioinformatics program Perseus for continued analysis and data preparation.